
各論
Ⅰ.遺伝性大腸癌の概要(Outlines of Hereditary Colorectal Cancer)
Ⅰ-1 基本的事項
Ⅰ-1-1 定義
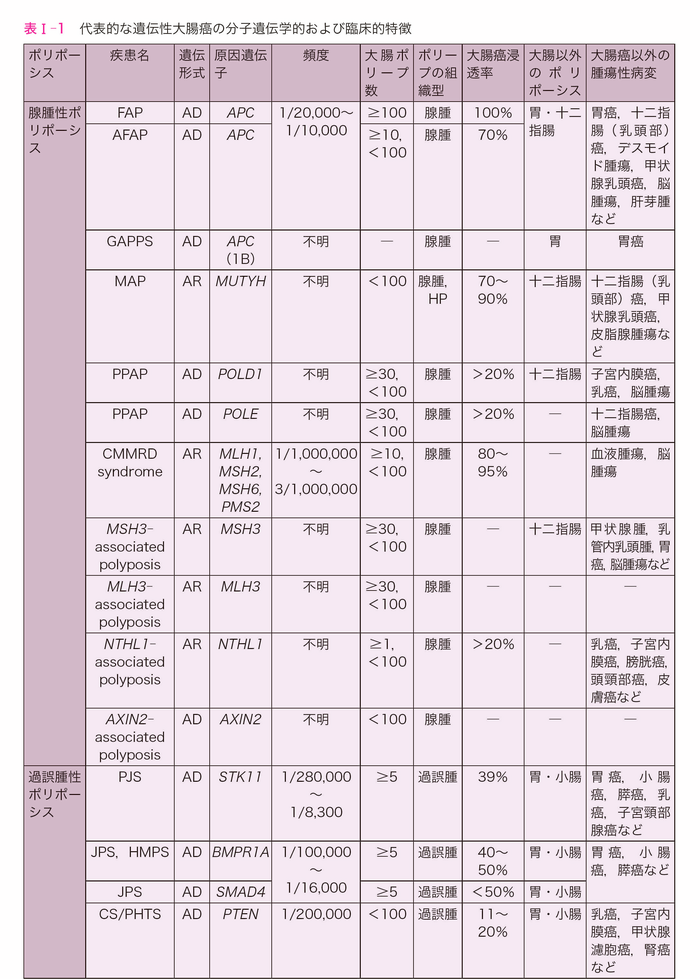
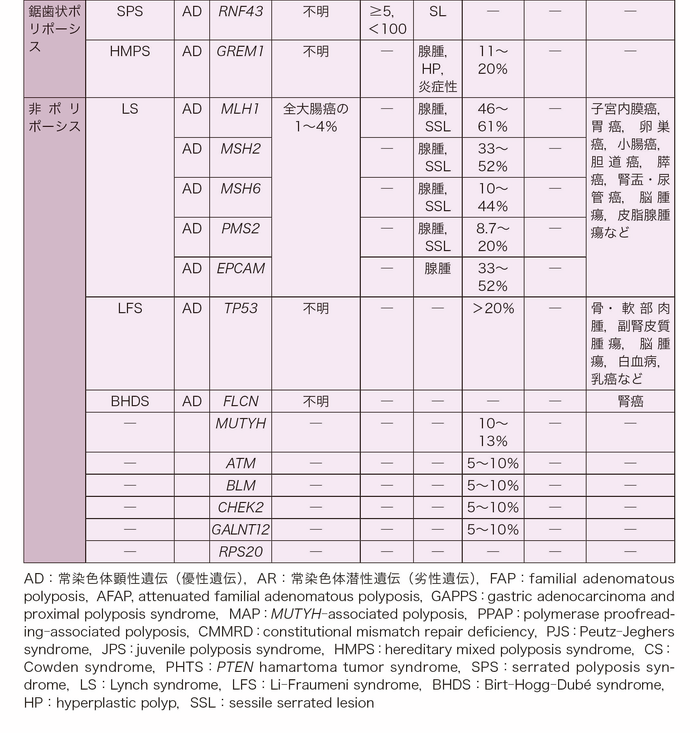
- 遺伝性大腸癌は大腸癌の発症リスクが高く原因遺伝子が同定されている疾患群を指し,原因遺伝子によって遺伝形式,大腸ポリープの密度,組織型,大腸癌発症リスク,関連する大腸外病変が異なる。代表的な遺伝性大腸癌の分子遺伝学的および臨床病理学的特徴を表Ⅰ-1にまとめた。
Ⅰ-1-2 分類
- 遺伝性大腸癌は,ポリポーシスの有無により大別できる。さらに,ポリポーシスを示す遺伝性大腸癌は,ポリープの組織型により,腺腫性ポリポーシス,過誤腫性ポリポーシス,鋸歯状ポリポーシスに分類できる。
- 腺腫性ポリポーシスは大腸ポリープ数が10個以上の腺腫性ポリープを認める患者を指す。大腸ポリープの数により密生型(1,000~2,000個以上),非密生型(100個以上1,000個未満),attenuated型(10個以上100個未満)に分類することがあるが,観察した年齢,使用した光学機器,色素散布や狭帯域光観察(narrow band imaging:NBI)を含めた観察方法,担当医により大腸ポリープ数の結果は異なることに注意が必要である。腺腫性ポリポーシスの中でも,数千個以上の腺腫性ポリープを認める場合は家族性大腸腺腫症(FAP)であることが多く,胃や十二指腸にもポリープが多発する。100個未満の場合は,FAP,MUTYH関連ポリポーシス(MUTYH-associated polyposis:MAP),ポリメラーゼ校正関連ポリポーシス(polymerase proofreading-associated polyposis:PPAP),先天性ミスマッチ修復欠損(constitutional mismatch repair deficiency:CMMRD)症候群,MSH3関連ポリポーシス(MSH3-associated polyposis),MLH3関連ポリポーシス(MLH3-associated polyposis),NTHL1関連ポリポーシス(NTHL1-associated polyposis)などが鑑別疾患として挙がるが,これらの腺腫性ポリポーシスでも大腸ポリープ数が100個を超えることがあるため,大腸ポリープ数のみで腺腫性ポリポーシスを鑑別することはできない。遺伝形式別では,APC,POLE,POLD1,AXIN2を原因遺伝子とする場合は常染色体顕性遺伝(優性遺伝)形式を,その他の腺腫性ポリポーシスは常染色体潜性遺伝(劣性遺伝)形式をとる。
- 過誤腫性ポリポーシス症候群は大腸のポリープ数が10個未満のことがある。代表的な過誤腫性ポリポーシスには,Peutz-Jeghers症候群(Peutz-Jeghers syndrome:PJS),若年性ポリポーシス症候群(Juvenile polyposis syndrome:JPS),Cowden症候群(Cowden syndrome:CS)/PTEN過誤腫症候群(PTEN hamartoma tumor syndrome:PHTS)があり,各疾患に特徴的な組織型を示す。過誤腫性ポリポーシスでは,大腸に加えて胃や小腸(十二指腸を含む)にもポリープが多発する傾向があり,口唇の色素斑や巨頭症など特徴的な外見を示すことがある1-6)。
- 鋸歯状ポリポーシスは,過形成性ポリープや鋸歯状病変などの多彩なポリープが発生する病態であり,この疾患群は鋸歯状ポリポーシス症候群(serrated polyposis syndrome:SPS)と総称される。鋸歯状ポリポーシス症候群の原因遺伝子について一定の見解は得られていないが,一部の患者にRNF43やGREM1,MUTYHの生殖細胞系列における病的バリアントが同定されたと報告されている(サイドメモⅠ-1:バリアント,生殖細胞系列バリアントと体細胞バリアント)7-9)。また,鋸歯状ポリープの発生には環境因子の関与も指摘されている。
- 大腸のポリープ数が少ない代表的な遺伝性大腸癌には,リンチ症候群とLi-Fraumeni症候群(Li-Fraumeni syndrome:LFS)がある。これらの疾患は外見上の特徴に乏しいため,臨床病理学的因子のみで診断することはできない。なお,リンチ症候群の原因遺伝子であるミスマッチ修復遺伝子の両アレルにおける病的バリアントを持つ常染色体潜性遺伝(劣性遺伝)性疾患に先天性ミスマッチ修復欠損(CMMRD)症候群があり,大腸に腺腫性ポリープが多発する(Ⅲ-2-2-4:先天性ミスマッチ修復欠損(CMMRD)症候群)。その他にも生殖細胞系列の病的バリアントが大腸癌の発症リスクと関連している遺伝子の報告がある。
Ⅰ-1-3 疫学
- 大腸癌患者の約30%は家族集積性があるか遺伝的素因がある10,11)(図Ⅰ-1)。遺伝性素因があるにもかかわらず家族集積性を伴わない例には,常染色体潜性遺伝(劣性遺伝)性疾患症例の場合や常染色体顕性遺伝(優性遺伝)性疾患でも体細胞モザイクや新生発端者(de novo症例)の場合がある。一方,家族集積性を伴うにもかかわらず原因遺伝子が同定されていない例には,家族性大腸癌タイプX(Ⅲ-2-2-5:家族性大腸癌タイプX)がある。
- 大腸癌患者を対象とした生殖細胞系列についてのマルチ遺伝子パネル検査(multi-genepanel testing, MGPT)によれば12,13),FAPが約0.5%,リンチ症候群が3%を占め,ATMやCHEK2,MUTYH,TP53に病的バリアントを持つ者も認められており,大腸癌全体の約5%が遺伝性大腸癌であった。また,遺伝性大腸癌以外にも遺伝性乳癌卵巣癌の原因遺伝子であるBRCA1,BRCA2がそれぞれ約1%,PALB2,BRIP1,NBN,RAD51Dなどが合わせて約3%に検出されており,これら大腸癌発症への関与が不明なものを含めると全大腸癌患者の約10%は遺伝性腫瘍であると見積もられる。
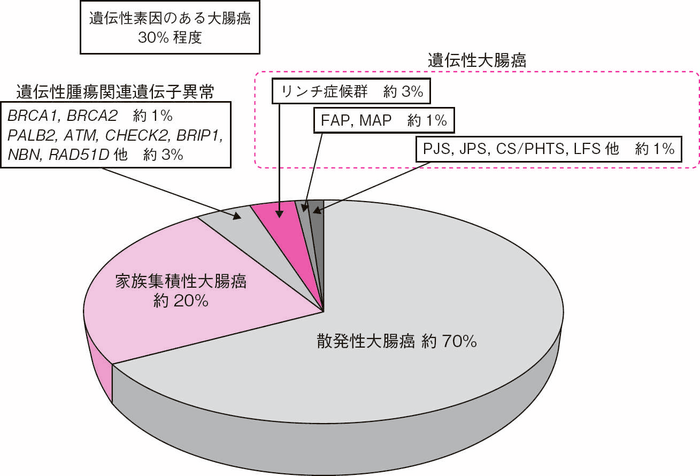
図Ⅰ-1 全大腸癌における遺伝性素因のある大腸癌の割合
Ⅰ-1-4 腫瘍発症リスク
- 遺伝性大腸癌における大腸癌の発症リスクは疾患により異なるものの,典型的FAPでは浸透率(サイドメモⅠ-1:浸透率)がほぼ100%であることを除けば,遺伝性大腸癌の原因遺伝子に病的バリアントを持つ者が必ずしも大腸癌を発症するとは限らない。また,リンチ症候群のように,同一の疾患でも原因遺伝子によって腫瘍の発症リスクが異なることがある14)。
- 遺伝性大腸癌では大腸癌以外にも発症リスクの高い腫瘍(病変)があり,それらは関連腫瘍(病変)と呼ばれる。FAPでは胃底腺ポリポーシスや十二指腸腺腫,リンチ症候群では婦人科腫瘍や泌尿器科腫瘍などが関連腫瘍(病変)であり,遺伝性大腸癌の関連腫瘍(病変)は臓器特異的に発生する。また,遺伝性大腸癌では大腸癌未発症のまま関連腫瘍(病変)が発生することもあり,リンチ症候群のおよそ35%の女性においては子宮内膜癌が初発がんである15,16)。したがって,遺伝性大腸癌にあたっては,消化器内科や消化器外科のみならず,診療科横断的な連携が重要である。
Ⅰ-1-5 発がんのメカニズム
- 現在までに同定されている遺伝性大腸癌の原因遺伝子はすべてがん抑制遺伝子である。APC,TP53,PTEN,SMAD4などは細胞の増殖を抑える遺伝子で,MLH1,MSH2,MUTYHなどはDNAの異常を修復する遺伝子である。したがって,遺伝性大腸癌では原因遺伝子の両アレルにおける機能喪失型バリアントが病的バリアントとなる。機能喪失型バリアントには,短縮型バリアントやLOH(loss of heterozygosity)(サイドメモⅠ-1:ヘテロ接合性の消失,染色体不安定性),エクソン単位での重複/欠失,一部のアミノ酸置換型バリアントがある。
- 常染色体顕性遺伝(優性遺伝)形式の遺伝性大腸癌では,原因遺伝子の生殖細胞系列の病的バリアントに野生型アレルにおける後天的な病的バリアントが加わると,同名蛋白質の機能が喪失し腫瘍(病変)が発生する17)。遺伝性大腸癌では生来すべての細胞において片アレルの原因遺伝子に病的バリアントを持つため,散発性大腸癌と比較して若年発症しやすく,多発しやすい(図Ⅰ-2)。
- FAPにおける大腸癌の発生は多段階発がんモデルで説明される18)。まず,大腸の上皮細胞においてAPC蛋白質の機能喪失により,細胞内に蓄積し核内移行が増加したβカテニンがTCF4と複合体を形成した結果,がん遺伝子などの転写が促進され,細胞増殖する。形態学的には異常腺窩巣(aberrant crypt foci:ACF)(サイドメモⅠ-1:異常腺窩巣)が発生すると考えられている19)。ACFから腺腫を経て大腸癌が発生するにはKRASやTP53など複数の遺伝子にも変化が加わるが,これには染色体不安定性(chromosomal instability:CIN)という遺伝子異常が起きやすい状態が関与していると考えられる(サイドメモⅠ-1:染色体不安定性)(図Ⅰ-3)。
- リンチ症候群における大腸癌の発生にはミスマッチ修復機構の破綻が関与している。細胞分裂に伴うDNAの複製は極めて正確に行われ,複製エラーの頻度は10-10~10-8である。万が一,DNAの複製エラーが生じても,リンチ症候群の原因遺伝子に由来するミスマッチ修復蛋白質がミスマッチや1~数塩基までの挿入/欠失のようなDNAの複製エラーを認識する。しかし,ミスマッチ修復遺伝子の生殖細胞系列に加えて後天的な病的バリアントが生じると,ミスマッチ修復機構は破綻しDNAの複製エラーを認識できなくなる。DNAの複製エラーはゲノム内の単純な反復配列であるマイクロサテライト領域に好発する。遺伝子産物(蛋白質)をコードする領域に反復配列を含む遺伝子もあり,その中には腫瘍抑制(TGFBR2など),細胞増殖,DNA修復(MSH3,MSH6など)やアポトーシス(BAXなど)などに関わる遺伝子もあり,ミスマッチ修復機構が破綻すると反復回数の異常(不安定性)が生じるためこれらの遺伝子の異常が蓄積し腫瘍が発生する。
- 常染色体潜性遺伝(劣性遺伝)形式の遺伝性大腸癌は両アレルにおける原因遺伝子の病的バリアントが原因であり,保因者である両親から病的バリアントをひとつずつ受け継いだ時に発症する。常染色体潜性遺伝(劣性遺伝)形式の遺伝性大腸癌の原因遺伝子はDNA修復遺伝子である。特定のDNA損傷が加わった場合にこれを修復することができないため腫瘍が発生する。
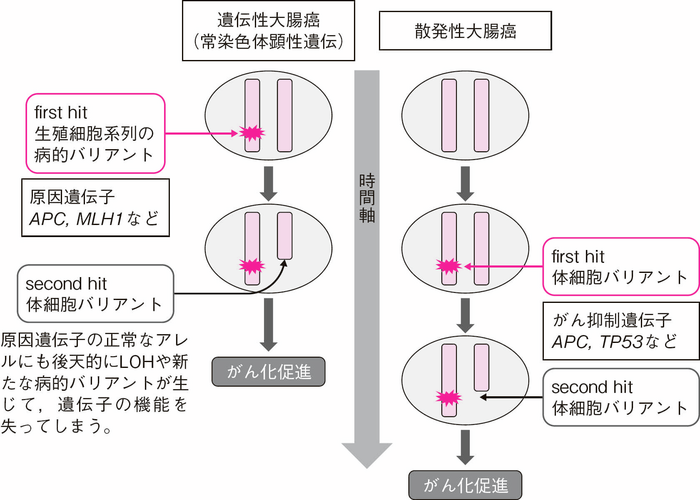
図Ⅰ-2 Knudsonによるがん抑制遺伝子のtwo-hit説によるがん化のメカニズム
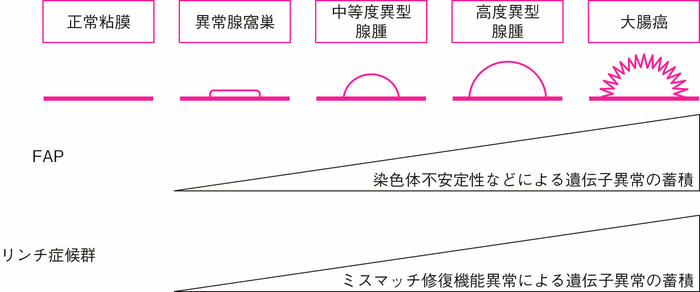
図Ⅰ-3 FAPとリンチ症候群の代表的ながん化のメカニズム
サイドメモI‒1
■バリアント
「バリアント」とは遺伝情報の多様性を意味する言葉で,主にDNAの塩基配列において参照配列と異なる塩基配列を指す。日本語訳として「多様体」が用いられることもあるが,「バリアント」のまま用いられることが多い。同様な言葉として「変異」があるが,生物学的意義を持たせた表現として使う場合とそうでない場合があるなど用い方に混乱がある。そのため,「変異(mutation)」という言葉はなるべく用いず「バリアント」を用い,生物学的意義や臨床的意義の評価を付加する場合は,pathogenic(病的)やbenign(病的でない),uncertain significance(意義不明)などの修飾語をつけて表現する。
■生殖細胞系列バリアントと体細胞バリアント
精子あるいは卵子を経由して受け継がれるDNAの塩基配列変化を生殖細胞系列バリアントという。受精卵の時点でその変化は存在するため,全身のすべての細胞に同じ変化が存在する。それに対して,身体を構成する生殖細胞以外の細胞(体細胞)に新たに生じた塩基配列の変化を体細胞バリアントという。
■浸透率
遺伝性疾患における原因遺伝子の遺伝型(Genotype)の保有者において病気が発症する確率である。100%の確率で発症する場合を完全浸透という。
■ヘテロ接合性の消失(loss of heterozygosity:LOH)
両親から各々受け継がれた遺伝情報のうち,相同な領域において異なる塩基配列が存在する場合をヘテロ接合性(heterozygosity)という。FAPの場合,正常細胞ではAPC遺伝子の片側にのみ病的なバリアントが存在しており,もう一方のAPC遺伝子は正常(野生型)である。この状態がヘテロ接合性であるが,がん化の過程で野生型のAPC遺伝子が欠失により失われることをLOHという。
■異常腺窩巣(aberrant crypt foci:ACF)
ACFは,内視鏡の通常観察では正常粘膜と区別することはできないが,拡大視観察ではメチレンブルーに濃染する異常腺管の集合として確認できる。ACFの一部は腺腫や癌の前駆病変と考えられている。
■染色体不安定性(chromosomal instability:CIN)
CINとは,がん細胞などで見られる染色体の数の異常や構造異常(欠失,重複,転座など)のこと。腫瘍化の原因になると考えられている。
Ⅰ-2 診断
Ⅰ-2-1 診断の意義
- 遺伝性大腸癌は遺伝学的検査により確定診断を行う(サイドメモⅠ-2:遺伝学的検査)。同じ表現型でも鑑別すべき疾患や原因遺伝子の候補は複数存在するため,表現型のみで診断することはできない。遺伝性大腸癌は原因遺伝子により,①遺伝形式,②悪性腫瘍の発症割合(浸透率),③併存疾患(関連腫瘍),④サーベイランス法が異なる。また,血縁者に対しても①血縁者診断,②リスク評価(再発率)に利用できる。したがって,遺伝学的検査による確定診断は,遺伝性大腸癌患者への適切な医療提供につながる。
- 遺伝学的検査・診断に際して,必要に応じて適切な時期に遺伝カウンセリングを実施する。遺伝カウンセリングは,情報提供だけではなく,患者・被検者等の自律的選択が可能となるような心理的社会的支援が重要であることから,当該疾患の診療経験が豊富な医師と遺伝カウンセリングに習熟した者が協力し,チーム医療として実施することが望ましいとされる20)。したがって,遺伝性大腸癌の検査および診断に際しては,疑われる疾患についての十分な情報と遺伝学的検査のメリット・デメリットについて予め提供することが重要である(Ⅰ-3:遺伝カウンセリング)。
サイドメモI‒2
■遺伝学的検査
「遺伝子検査」という用語は,「体細胞の遺伝子検査」なのか「生殖細胞系列の遺伝子検査」なのか区別がつかないため,前者を「体細胞遺伝子検査」,後者を「遺伝学的検査」と呼ぶことが日本臨床検査標準協議会の「遺伝子関連検査標準化専門委員会」から提言された。また,これらの呼称は,遺伝医学関連学会などにより作成された日本医学会の「医療における遺伝学的検査・診断に関するガイドライン」20)でも分類・定義されている。
Ⅰ-2-2 診断の流れ
STEP 1 臨床情報によるリスク評価
- 遺伝性大腸癌の診断は臨床情報から遺伝性大腸癌のリスク評価を行うことから始まる。日常臨床において,遺伝性大腸癌であるかどうかの手掛かりは,①若年発症の有無,②同時性・異時性の大腸癌の発生,③大腸癌以外の併存疾患の有無,④身体的特徴,⑤第3度近親者(少なくとも第2度近親者)までの家族歴の有無を確認することにある(図Ⅰ-4)。
遺伝性大腸癌では大腸以外にも病変が発生することが多く(Ⅰ-1-4:腫瘍発症リスク),歯牙の異常や口唇の色素斑,巨頭症などの身体的特徴を示すものがあり,これらの所見は診断の補助や原因遺伝子の推定に利用することができる。核家族化や親族間交流の減少により家族歴の聴取においては第2度近親者の情報さえ得られないこともあるが,父方と母方を区別して家系図を作成することで遺伝性大腸癌の遺伝形式を推定することができる(付録Ⅲ:家系図の記載法)。特に,ポリポーシスを認めない遺伝性大腸癌では外見上の特徴に乏しいため,関連腫瘍発生の有無は遺伝性大腸癌スクリーニングの鍵となる。 - 遺伝性大腸癌の診断において消化管内視鏡検査所見は極めて重要である。そこで,大腸(下部消化管)内視鏡検査だけでなく上部消化管内視鏡検査においても①ポリープの数,②ポリープの種類,③ポリープの局在を確実に記録する。
- この臨床情報によるリスク評価で遺伝性大腸癌が疑われる場合にはSTEP 2へと進む。なお,家系内に既に遺伝性大腸癌と診断されている者がいる大腸癌患者が当該疾患に合致する表現型を認める場合にはSTEP 3へと進む21)。
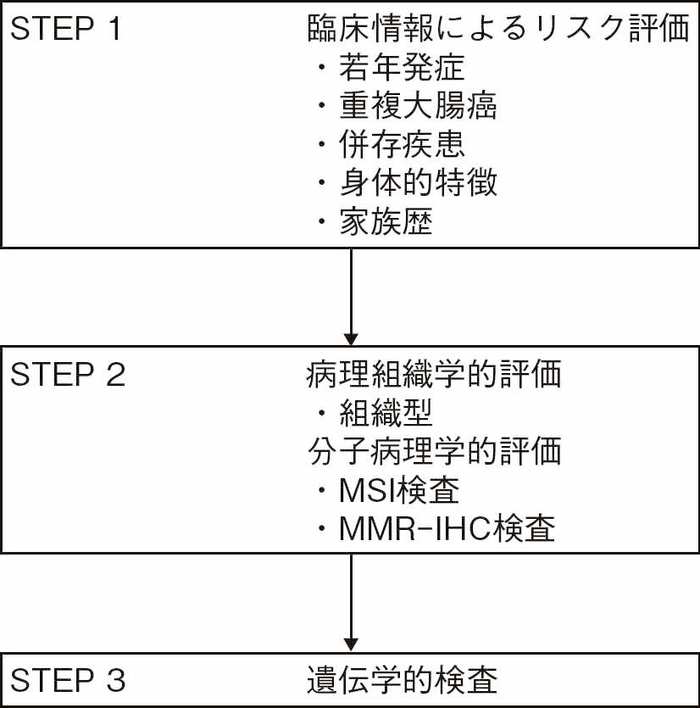
図Ⅰ-4 遺伝性大腸癌のリスク評価と診断の流れ
MSI検査:マイクロサテライト不安定性検査,
MMR-IHC検査:ミスマッチ修復蛋白質の免疫組織化学検査
STEP 2 病理組織学的および分子病理学的評価
- STEP 1で大腸にポリポーシスを認めた場合には生検を実施し,病理組織学的評価により組織型を確認する。図Ⅰ-5 と表Ⅰ-2に病理組織学的所見に基づいた遺伝性大腸ポリポーシスの診断のフローと鑑別すべき疾患についてまとめた。各ポリポーシスには複数の鑑別すべき疾患と候補となる原因遺伝子がある。
- STEP 1でポリープ数が少ない場合には,大腸癌組織を用いてMSI検査またはミスマッチ修復蛋白質の免疫組織化学検査を行う(各論Ⅲ:リンチ症候群)。リンチ症候群に発生した大腸癌のほとんどは,MSI-Highまたは原因遺伝子に対応したミスマッチ修復蛋白質の発現消失を認める。本人や血縁者においてリンチ症候群関連腫瘍の発症が多い場合には,これらの結果にかかわらずSTEP 3へと進む。
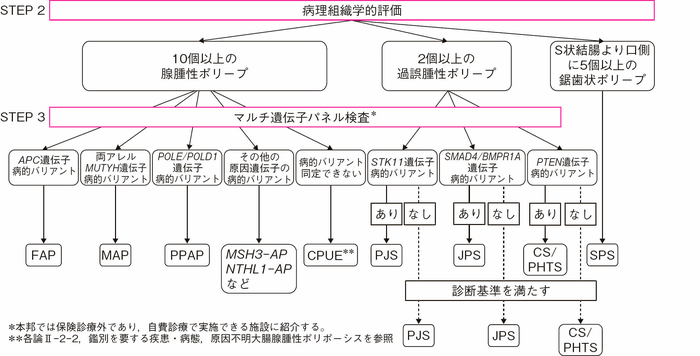
図Ⅰ-5 遺伝性大腸ポリポーシス 診断のフローチャート
FAP:familial adenomatous polyposis, MAP:MUTYH-associated polyposis, PPAP:porymerase proofreading-associated polyposis, MSH3-AP:MSH3-associated polyposis, NTHL1-AP:NTHL1-associated polyposis, CPUE:colonic adenomatous polyposis of unknown etiology, PJS:Peutz-Jeghers syndrome, JPS:juvenile polyposis syndrome, CS/PHTS:Cowden syndrome/PTEN hamartoma tumor syndrome, SPS:serrated polyposis syndrome

STEP 3 遺伝学的検査
- 遺伝性大腸癌の確定診断は遺伝学的検査によって行われる。通常,採血管1~2本分の血液からDNAを抽出し,生殖細胞系列の遺伝子異常の有無について解析する。遺伝学的検査による原因遺伝子の同定は,発がんリスクを推定することができるため,患者への情報提供やサーベイランスに利用できる22,23)他,未発症者を含む血縁者の診断(血縁者診断)に利用することができる。
- 遺伝性大腸癌の遺伝学的検査には,①ダイレクトシークエンス法,②Multiplex Ligation-dependent Probe Amplification(MLPA)法,③MGPTによるターゲットシークエンス法,④全エクソームシークエンス法,⑤全ゲノムシークエンス法がある(表Ⅰ-3)。
- ①のダイレクトシークエンス法では推定される原因遺伝子の蛋白質をコードするエクソンとエクソン-イントロン境界領域の塩基配列を一塩基ずつ確認することができる。②のMLPA法ではエクソン毎に固有の標識を付加することで,エクソン単位の欠失や重複を確認することができる。原因遺伝子が高い確率で推定されている場合,遺伝学的検査はダイレクトシークエンス法やMLPA法が用いられる。ただし,数千個の腺腫性ポリープを認める場合でもAPCに病的バリアントが認められるとは限らない。また,血縁者診断では発端者に認められた病的バリアントのみ(シングルサイト)の解析でもよいため,検出目的のバリアントに応じてダイレクトシークエンス法あるいはMLPA法が用いられる。③のターゲットシークエンス法では,疑われる遺伝性大腸癌の原因遺伝子をターゲットとしたパネルを作成し,次世代シークエンサーを用いて解析する。遺伝性大腸癌では臨床所見から複数の原因遺伝子を推定することが多いため,最近ではMGPTによるターゲットシークエンス法が行われている。腺腫性ポリポーシスではAPC, MUTYH, POLD1, POLEなどの遺伝子を,リンチ症候群ではMLH1, MSH2, MSH6, PMS2, EPCAMを解析する必要があるが,ターゲットシークエンス法では複数の遺伝子を同時に解析することが可能である。したがって,MGPTにBRCA1/2を加えた場合には遺伝性乳癌卵巣癌と診断される症例もある24,25)。④の全エクソームシークエンス法では,全ての遺伝子のエクソンとエクソン-イントロン境界領域について,⑤の全ゲノムシークエンス法では,非コード領域のイントロンを含めたすべての領域について次世代シークエンサーを用いて解析する。全エクソームシークエンス法や全ゲノムシークエンス法では,すべての遺伝子を解析するため,腫瘍の発生に関連がないとされる遺伝性疾患を診断することがある。また,全ゲノムシークエンス法では,他の解析方法では検出しにくいイントロン領域の異常や転座,逆位などの遺伝子異常が検出できる。
- 遺伝性大腸癌の体細胞モザイク症例(Ⅱ-2-2:鑑別を要する疾患・病態)では,身体の一部の細胞のみが病的バリアントを持つため,病的バリアントの頻度が低く,ダイレクトシークエンス法の検出感度を下回ることがある。しかし,次世代シークエンサーを用いた解析では低い頻度のバリアントを検出することができるため,体細胞モザイクの診断に有用である。
- 50歳未満の大腸癌患者にMGPTによるターゲットシークエンス法を行った結果,約20%の患者に生殖細胞系列の病的バリアントが認められており26),NCCNガイドライン(Version2. 2023)14)では50歳未満の全大腸癌患者と50歳以上でもMSI-High/dMMRであればMGPTによるターゲットシークエンスの実施を推奨している。
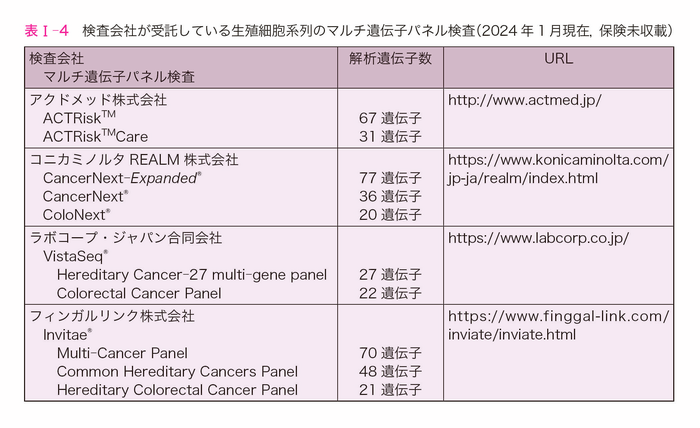
- 以上より,遺伝性大腸癌が疑われる症例に対してはMGPTによるターゲットシークエンス法を用いた遺伝学的検査を実施することが望ましい。しかし,本邦では遺伝性大腸癌に対する遺伝学的検査は製造販売承認も保険承認も受けていないため,自費診療または研究目的での実施となる(2024年1月現在)。現在,遺伝性腫瘍のMGPTによるターゲットシークエンスが可能な検査会社は表Ⅰ-4の通りである。


Ⅰ-2-3 がんゲノムプロファイリング検査における生殖細胞系列所見(二次的所見)
- 標準治療がない固形がん(原発不明がんや希少がんなど),または標準治療が終了となった固形がん(終了が見込まれる者を含む)患者を対象として,推奨薬探索目的に(包括的)がんゲノムプロファイリング検査(表Ⅰ-5)が行われた際に,生殖細胞系列所見(二次的所見)として遺伝性大腸癌と診断あるいは疑われる場合がある。
- OncoGuideTM NCCオンコパネルシステムとGenMineTOPがんゲノムプロファイリングシステムでは血液中の有核細胞由来のDNAを正常コントロールとして解析するため,正常細胞由来の検体の解析結果は遺伝学的検査の結果と同様に扱う。したがって,正常細胞由来の検体の解析で認められた病的バリアントは,生殖細胞系列病的バリアント(germline pathognic variant, GPV)となる。一方,FoundationOne® CDxがんゲノムプロファイルとFoundationOne® Liquid CDxがんゲノムプロファイル,Guardant360® CDxがん遺伝子パネルでは腫瘍組織のみを解析するため,その中に含まれるDNAの多くは腫瘍細胞由来である。したがって,腫瘍組織のみの解析で生殖細胞系列由来の可能性がある病的バリアント(presumed germline pathogenic variant, PGPV)が認められた場合,遺伝性大腸癌の確認検査として遺伝学的検査が必要となる。なお,がんゲノムプロファイリング検査で遺伝性大腸癌が疑われる場合には,「がん遺伝子パネル検査二次的所見患者開示推奨度別リスト」を参考にする27)。
Ⅰ-2-4 遺伝学的検査結果の解釈
- 検出されたバリアントの臨床的意義については,ClinVar28)やInSiGHT29)のクラス分類(表Ⅰ-6)等で評価する。
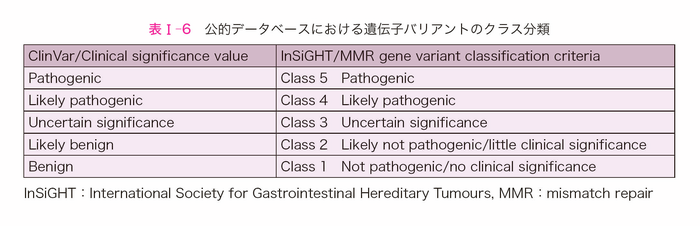
A)「病的バリアントもしくは病的バリアントの可能性が高い」場合
―pathogenicまたはlikely pathogenicに相当
- 遺伝性疾患として医学的管理を行う。ただし,FAPのような浸透率がほぼ100%である疾患を除き,未発症の病的バリアント保持者が一生涯に必ずがんを発症するとは限らないことも理解してもらう。
B)「意義不明バリアント」が検出された場合
―uncertain significanceに相当
- 疾患への影響が分からない遺伝子変化のことを意義不明バリアント(variants of uncertain significance, VUS)として報告される。例えば,塩基配列が一つ変化してもアミノ酸合成には影響しないサイレントバリアントや,アミノ酸の置換が起きるミスセンスバリアントが疾患発症に影響するのか判断できない場合である。この場合,このバリアントの意義が証明されるまで,次項の「遺伝子異常が検出されない」と同様に対応することがすすめられる。
C)「遺伝子異常が検出されない」場合
―benignまたはlikely benignに相当
〔家系内で遺伝子異常が確定している場合〕
- 遺伝学的検査で確定診断されている家系において,同じ遺伝子変化が認められなかった場合は,家系で認められた遺伝性疾患ではないと判断する。その場合でも,一般集団の発がんリスクがあることは理解してもらう。
〔家系内の確定診断がされていない場合〕
- 遺伝学的検査に用いられた方法によっては検出できないバリアントを有しているか,あるいは未知の原因遺伝子の異常によることなども考えられ,慎重に対応する。例えば,臨床的にポリープ数が100~1,000個ほどあるFAPでも,APCの病的バリアントの検出率は60%程度30)と100%には至らないことから,技術的な問題で未知のバリアントが検出されていないことやAPC以外の遺伝子の異常の可能性も考えなければならない。臨床学的に遺伝性疾患と考えられる場合,遺伝学的検査で病的バリアントが検出されなくても,実地臨床では遺伝性疾患と同様に対応していくことが望ましい。
Ⅰ-3 遺伝カウンセリング
Ⅰ-3-1 定義
- 遺伝カウンセリングは,疾患の遺伝学的関与について,その医学的影響,心理学的影響および家族への影響を人々が理解し,それに適応していくことを助けるプロセスである。このプロセスには,1)疾患の発生および再発の可能性を評価するための家族歴および病歴の解釈,2)遺伝現象,検査,マネージメント,予防,資源および研究についての教育,3)インフォームド・チョイス(十分な情報を得た上での自律的選択),およびリスクや状況への適応を促進するためのカウンセリング,などが含まれる20)。
Ⅰ-3-2 リスクコミュニケーションと意思決定支援
- 遺伝カウンセリングでは,遺伝性腫瘍のリスクをアセスメントし,その情報を患者・家族が正確に理解できるようコミュニケーションの中で,情報提供していくことが重要となる。このような,リスクコミュニケーションは,患者・家族が遺伝学的情報を活用して,がんのリスクを軽減し,生活の質を改善するためには重要となる。
- 遺伝カウンセリングの中で,患者・家族が抱える心理・社会的問題が表面化した時,その問題をどのように解決していくのか,意思決定が必要となる。共有型意思決定(shared decision making:SDM)とは,患者が支援を受けながら意思決定に関わり,意思決定ニーズを満たし,患者と2名以上の医療従事者が合意する質の高い意思決定を成し遂げるプロセスであり,以下のステップが示されている31)。
(1) 意思決定すべき事柄を明確にする。 (2) 選択肢,利益及び不利益に関する情報を交換する。 (3) 患者が最も得たいと思っている利益や最も回避すべき不利益を表現するための援助を行う。 (4) 選択肢の実施可能性を探る。 (5) 実際に選択する選択肢について合意するために優先順位について話し合う。 - 心理・社会的問題は多義にわたり,複雑性を伴うことから,問題の性質に応じて複数の医療従事者が関わる中で,意思決定支援を進めていく必要がある。
Ⅰ-3-3 心理社会的問題
- 遺伝カウンセリング対象者が抱える心理社会的問題について,以下の6項目(表Ⅰ-7)が示されている32)。
- 遺伝カウンセリングでは,このような問題をできるだけ早い段階で抽出し,患者および家族がこれらの問題に向き合い,乗り越えていくことができるよう,チーム医療の中で支援していく必要がある。

Ⅰ-3-4 手法
- 遺伝カウンセリングでは,対話を通じて,遺伝性疾患や遺伝学的検査に関する正確な情報をクライエントに提供し,疑問に適切に答えることによって,クライエントの理解を深め,不安や悩みにこたえることによって,今後の生活に向けて自らの意思で選択し,行動できるように支援する。すなわち,正確な遺伝医学の知識をわかりやすく伝えることにより,遺伝的問題で悩む患者家族の不安を取り除く。また,実際の遺伝カウンセリングにあたっては,①相手をそのまま受け入れる受容的態度,②非指示的対応(態度),③共感的理解で臨む。受容的態度とは,相手をそのまま,否定も肯定もせず,評価を加えずに受け入れることで,非指示的対応とは,相手の話しを注意深く,正確に,真摯的に傾聴することで,共感的理解とは,価値観の違う相手の立場になって理解しようとする態度である。なお,クライエントの考え方,感受性,事前の知識,理解力,不安の大きさ,医療に対する信頼感が個々で異なることに注意する。
- 遺伝カウンセリングに関する基礎知識・技能については,全ての医師が習得しておくことが望ましいとされ,遺伝学的検査・診断を担当する医師および医療機関は,必要に応じて,医師・非医師の専門家による遺伝カウンセリングを提供するか,または紹介する体制を整えておく必要がある20)。
Ⅰ-3-5 情報収集内容
- 遺伝カウンセリングでは,受診のきっかけとなった遺伝性腫瘍の情報と家系情報を確認し,リスク評価を正確に行う。さらに,遺伝学的検査,健康管理,サーベイランスなどの認識・価値観・意向を確認しながら,当事者に必要な情報提供に繋げる(表Ⅰ-8)。

Ⅰ-3-6 情報提供内容
- クライエントが遺伝性大腸癌について正確に理解するために,がんと遺伝に関するさまざまな情報を提供する必要がある(表Ⅰ-9)。また,遺伝学的検査を行う際に結果によって本人や家族にもたらす心理的影響や社会的差別への配慮などにも留意する。
- 遺伝学的検査を施行する前にその臨床的意義について説明し,検査によるメリットとデメリット(表Ⅰ-10)について理解していることが必須である。
- 遺伝学的検査は,医学的,倫理的,経済的,技術的なさまざまな観点でクライエントの負担にならないように配慮しながら,文章による検査の説明と同意書を作成し,インフォームド・コンセントを受けたうえで実施する。遺伝学的検査を受ける前後だけではなく,必要に応じて遺伝カウンセリングを継続する。
- 遺伝学的検査の結果開示時には,家族の同席について希望の有無を確認する。同席を希望しない場合,個別に時間と場所を確保する。
サイドメモI-3
■サーベイランス上の注意
遺伝性大腸癌の随伴病変や関連腫瘍を含めたサーベイランスには,大腸内視鏡検査や子宮内膜組織診など,不快感や苦痛などをともなうものがある。前処置を含めた検査にともなう負担は,サーベイランスの間隔の遅延や遵守率の低下につながる可能性があるため,不快感や苦痛を和らげるための配慮をする。


Ⅰ-3-7 導入のタイミング
- 遺伝性大腸癌に関連する遺伝カウンセリングを提供するタイミングとしては,以下の3つが想定される。
- 臨床所見(発症年齢,重複癌/多発癌,家族歴,病理組織学的所見など)から遺伝性大腸癌を疑う場合
- コンパニオン診断(MSI検査またはMMR-IHC検査)でMSI-HighまたはdMMRを認めた場合
- 包括的がんゲノムプロファイリング検査で遺伝性腫瘍が診断または疑われた場合
- 遺伝学的検査を実施して病的バリアントの結果が得られた場合には,at-riskとされる家系員への遺伝カウンセリング(Ⅱ-4:家族(血縁者)・小児への対応,Ⅲ-5:家族(血縁者)への対応)へ繋げていくこと,サーベイランスの観点から継続的な遺伝カウンセリングを実施することが重要である。
Ⅰ-3-8 未成年への対応
- 日本医学会「医療における遺伝学的検査・診断に関するガイドライン」によれば,一般に成年期以降に発症する疾患の発症前遺伝学的検査など,未成年のうちに遺伝学的検査を実施することにメリットが多くない場合は,本人が成人し,自律的に判断できるようになるまで実施を延期すべきだが,早期診断により予防や早期治療が可能となるような場合には,両親などから代諾を得,また本人にも理解度に応じた説明を行い,了解(インフォームド・アセント)を得てから実施することが望まれる20)。(Ⅱ-4:家族(血縁者)・小児への対応,Ⅲ-5:家族(血縁者)への対応参照)
Ⅰ-3-9 遺伝学的検査・診断に関するガイドラインおよび指針
- 遺伝学的検査の実施時には,以下のガイドラインや指針を遵守する20,33,34)。また,被検者のプライバシーに配慮し,記録の保管は慎重に対処する。
- 日本医学会「医療における遺伝学的検査・診断に関するガイドライン(2022年3月改定)」
- 日本遺伝性腫瘍学会「家族性腫瘍における遺伝学的検査の研究とこれを応用した診療に関する指針(2019年版)」
- 文部科学省・厚生労働省・経済産業省「人を対象とする生命科学・医学系研究に関する倫理指針(令和5年3月27日一部改正)」
- 「ゲノム医療における情報伝達プロセスに関する提言―その1:がん遺伝子パネル検査を中心に(改定第2版)」
- 「ゲノム医療における情報伝達プロセスに関する提言―その2:次世代シークエンサーを用いた生殖細胞系列網羅的遺伝学的検査における具体的方針(改定版)」
- がん遺伝子パネル検査の実施時には,ゲノム医療を実施する際の患者・家族への説明事項や留意事項を生殖細胞系列所見(二次的所見)への対応を含めてまとめられた以下の提言(国立研究開発法人 日本医療研究開発機構(AMED)のゲノム創薬基盤推進研究事業)などを遵守する35,36)。
- 「ゲノム医療における情報伝達プロセスに関する提言―その1:がん遺伝子パネル検査を中心に(改定第2版)」
- 「ゲノム医療における情報伝達プロセスに関する提言―その2:次世代シークエンサーを用いた生殖細胞系列網羅的遺伝学的検査における具体的方針(改定版)」
Ⅰ-3-10 遺伝性腫瘍診療における遺伝カウンセリング体制
- 遺伝カウンセリングの役割は,遺伝学的検査に関わる情報提供に留まらず,患者の自律的選択,生活調整,心理的社会的支援など,クライエントがサーベイランスを含めた包括的なケアが受けられるよう,多職種・他部門間で連携を図ることも重要な役割といえる。そのためには,下記に示した専門職が,各診療科医師,看護師,臨床心理士,医療ソーシャルワーカー,臨床検査技師等とチームで医療体制を整備していく必要がある。
- 現在,本邦における遺伝医療の専門職に係る認定制度には以下のものがある。
- 臨床遺伝専門医制度
http://www.jbmg.jp/ - 遺伝性腫瘍専門医制度
https://jsht-info.jp/medical_personnel/specialist/fcc/specialist01.html - 認定遺伝カウンセラー制度
http://plaza.umin.ac.jp/~GC/ - 家族性腫瘍カウンセラー制度
https://jsht-info.jp/medical_personnel/specialist/fcc/counselor.html - 遺伝性腫瘍コーディネーター制度
https://jsht-info.jp/medical_personnel/specialist/fcc/coordinator.html - 遺伝看護専門看護師
https://www.nurse.or.jp/nursing/qualification/vision/cns/index.html
- 臨床遺伝専門医制度
文献
- 山本博徳,阿部孝,石黒信吾,他: 小児・成人のためのPeutz-Jeghers 症候群診療ガイドライン(2020年版).遺伝性腫瘍2020; 20: 59-78.
- 松本主之,新井正美,岩間達,他: 小児・成人のための若年性ポリポーシス症候群診療ガイドライン(2020年版).遺伝性腫瘍2020; 20: 79-92.
- 高山哲治,五十嵐正広,大住省三,他: 小児・成人のためのCowden 症候群/PTEN 過誤腫症候群診療ガイドライン(2020 年版).遺伝性腫瘍2020; 20: 93-114.
- Yamamoto H, Sakamoto H, Kumagai H, et al.: Clinical Guidelines for Diagnosis and Management of Peutz-Jeghers Syndrome in Children and Adults. Digestion 2023; 104: 335-347.PMID: 37054692]
- Matsumoto T, Umeno J, Jimbo K, et al.: Clinical Guidelines for Diagnosis and Management of Juvenile Polyposis Syndrome in Children and Adults-Secondary Publication. J Anus Rectum Colon 2023; 7: 115-125.[PMID:37054692]
- Takayama T, Muguruma N, Igarashi M, et al.: Clinical Guidelines for Diagnosis and Management of Cowden Syndrome/PTEN Hamartoma Tumor Syndrome in Children and Adults-Secondary Publication. J Anus Rectum Colon 2023; 7: 284-300.[ PMID: 37900693]
- Gala MK, Mizukami Y, Le LP, et al.: Germline mutations in oncogene-induced senescence pathways are associated with multiple sessile serrated adenomas. Gastroenterology 2014; 146: 520-529.[PMID: 24512911]
- Jaeger E, Leedham S, Lewis A, et al.: Hereditary mixed polyposis syndrome is caused by a 40-kb upstream duplication that leads to increased and ectopic expression of the BMP antagonist GREM1. Nat Genet 2012; 44: 699-703.[PMID: 22561515]
- Buchanan D, Young J: A Perspective on Biallelic MUTYH Mutations in Patients with Hyperplastic Polyposis Syndrome. Gastroenterology 2009; 136: 2407-2408.[PMID: 19406141]
- Macaron C, Leach BH, Burke CA: Hereditary colorectal cancer syndromes and genetic testing. J Surg Oncol 2015; 111: 103-111.[PMID: 24975382]
- Hampel H, Frankel WL, Martin E, et al.: Screening for the Lynch syndrome(hereditary nonpolyposis colorectal cancer). N Engl J Med 2005; 352: 1851-1860.[PMID: 15872200]
- Yurgelun MB, Kulke MH, Fuchs CS, et al.: Cancer Susceptibility Gene Mutations in Individuals With ColorectalCancer. J Clin Oncol 2017; 35: 1086-1095.[PMID: 28135145]
- Uson PLS Jr, Riegert-Johnson D, Boardman L, et al.: Germline Cancer Susceptibility Gene Testing in UnselectedPatients With Colorectal Adenocarcinoma: A Multicenter Prospective Study. Clin Gastroenterol Hepatol 2022; 20:e508-e528.[PMID: 33857637]
- National Comprehensive Cancer Network: NCCN Clinical Practice Guidelines in Oncology: Genetic/Familial High-Risk Assessment: Colorectal. Version 2. 2023. Available from https://www.nccn.org/guidelines/guidelines-detail?category=2&id=1436
- Møller P, Seppälä T, Bernstein I, et al.: Cancer incidence and survival in Lynch syndrome patients receiving colonoscopic and gynaecological surveillance: first report from the prospective Lynch syndrome database. Gut 2017; 66: 464-472.[PMID: 26657901]
- Saita C, Yamaguchi T, Horiguchi SI, et al.: Tumor development in Japanese patients with Lynch syndrome. PLoS One 2018; 13: e0195572.[ PMID: 29672549]
- Knudson AG Jr: Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci U S A 1971; 68: 820-823.[ PMID: 5279523]
- Vogelstein B, Fearon ER, Hamilton SR, et al.: Genetic alterations during colorectal-tumor development. N Engl J Med 1988; 319: 525-532.[PMID: 2841597]
- Takayama T, Ohi M, Hayashi T, et al.: Analysis of K-ras, APC, and beta-catenin in aberrant crypt foci in sporadic adenoma, cancer, and familial adenomatous polyposis. Gastroenterology 2001; 121: 599-611.[PMID: 11522744]
- 日本医学会:「医療における遺伝学的検査・診断に関するガイドライン(2022年3月改定)」Avialable from https://jams.med.or.jp/guideline/index.html
- Kiyozumi Y, Matsubayashi H, Horiuchi Y, et al.: Germline mismatch repair gene variants analyzed by universal sequencing in Japanese cancer patients. Cancer Med 2019; 8: 5534-5543.[PMID: 31386297]
- Sinha A, Tekkis PP, Gibbons DC, et al.: Risk factors predicting desmoid occurrence in patients with familial adenomatous polyposis: a meta-analysis. Colorectal Dis 2011; 13: 1222-1229.[PMID: 20528895]
- Chenbhanich J, Atsawarungruangkit A, Korpaisarn S, et al.: Prevalence of thyroid diseases in familial adenomatous polyposis: a systematic review and meta-analysis. Fam Cancer 2019; 18: 53-62.[PMID: 29663106]
- Susswein LR, Marshall ML, Nusbaum R, et al.: Pathogenic and likely pathogenic variant prevalence among the first 10,000 patients referred for next-generation cancer panel testing. Genet Med 2016; 18: 823-832.[PMID:26681312]
- Yurgelun MB, Allen B, Kaldate RR, et al.: Identification of a Variety of mutations in cancer predisposition genes in patients with suspected Lynch syndrome. Gastroenterology 2015; 149: 604-613.e20.[PMID: 25980754]
- Stoffel EM, Koeppe E, Everett J, et al.: Germline genetic features of young individuals with colorectal cancer. Gastroenterology 2018 Mar; 154(4): 897-905.e1.[PMID: 29146522]
- がんゲノム医療中核拠点病院等連絡会議 二次的所見ワーキンググループ(SFWG): 「がん遺伝子パネル検査二次的所見患者開示推奨度別リスト(Ver4.2_20231003)」Available from https://www.ncc.go.jp/jp/c_cat/jitsumushya/030/Potentially_Actionable_SF_Gene_List_Ver4.2_20231003.pdf
- Landrum MJ, Lee JM, Riley GR, et al.: ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res 2014; 42: D980-D985.[PMID: 24234437]
- Thompson BA, Spurdle AB, Plazzer JP, et al.: Application of a 5-tiered scheme for standardized classification of 2,360 unique mismatch repair gene variants in the InSiGHT locus-specific database. Nat Genet 2014; 46: 107-115.[PMID: 24362816]
- Grover S, Kastrinos F, Steyerberg EW, et al.: Prevalence and phenotypes of APC and MUTYH mutations in patients with multiple colorectal adenomas. JAMA 2012; 308: 485-492.[PMID: 22851115]
- Légaré F, Stacey D, Gagnon S, et al.: Validating a conceptual model for an inter-professional approach to shared decision making: a mixed methods study. J Eval Clin Pract 2011; 17: 554-564.[PMID: 20695950]
- Eijzenga W, Hahn DE, Aaronson NK, et al.: Specific psychosocial issues of individuals undergoing genetic counsel ing for cancer―a literature review. J Genet Couns 2014; 23: 133-146.[PMID: 23996531]
- 日本遺伝性腫瘍学会: 「家族性腫瘍における遺伝学的検査の研究とこれを応用した診療に関する指針(2019年版)」Available from http://jsft.umin.jp/information/opinion/index.html(2024/4/1)
- 文部科学省・厚生労働省・経済産業省:「人を対象とする生命科学・医学系研究に関する倫理指針(令和 5年3月27日一部改正)」Available from https://www.mhlw.go.jp/stf/seisakunitsuite/bunya/hokabunya/kenkyujigyou/i-kenkyu/index.html(2024/4/1)
- 日本医療研究開発機構: 「ゲノム医療における情報伝達プロセスに関する提言―その1: がん遺伝子パネル検査を中心に(改定第2版)」Available from https://www.amed.go.jp/news/seika/kenkyu/20200121.html(2024/4/1)
- 日本医療研究開発機構: 「ゲノム医療における情報伝達プロセスに関する提言―その2: 次世代シークエンサーを用いた生殖細胞系列網羅的遺伝学的検査における具体的方針(改定版)」Available from https://www.amed.go.jp/news/seika/kenkyu/20200121.html(2024/4/1)









お問い合わせ・事務局
〒102-0075東京都千代田区三番町2 三番町KSビル
| 電話 | 03-3263-8697 |
| FAX | 03-3263-8687 |
| jsccr@secretariat.ne.jp | |
| 業務時間 | 平日9時-18時 |